
Clinical trials represent the most critical phase of drug development, where experimental medicines are tested in humans to establish safety, efficacy, and therapeutic value. Governed by strict ethical and scientific standards, clinical trials ensure that patient welfare remains central while generating reliable data for regulatory approval. From protocol development and ethics committee oversight to pharmacovigilance and ongoing monitoring, every stage of a clinical trial is closely regulated across the globe.
Download UNIT 4 – Clinical Trials Notes
Get simplified revision notes for this unit:
⬇️
Download Unit 4 Notes PDF
Importance of Clinical Trials
Clinical trials bridge the gap between laboratory research and real-world medical practice. They provide scientific evidence on how a drug behaves in the human body, its therapeutic benefits, and potential risks. Regulatory authorities rely heavily on clinical trial data to decide whether a drug can be approved for marketing. As a result, trials must be designed and conducted with utmost rigor, transparency, and ethical responsibility.
Developing Clinical Trial Protocols
Foundation of a Well-Designed Trial
A clinical trial protocol is a detailed written plan that describes how a trial will be conducted. It defines the study objectives, trial design, methodology, inclusion and exclusion criteria, dosing schedules, endpoints, and statistical considerations. A well-structured protocol ensures consistency across study sites and minimizes bias, thereby improving data credibility.
Protocols are developed in accordance with international guidelines such as Good Clinical Practice (GCP). Before implementation, they must be reviewed and approved by regulatory authorities and ethics committees to ensure scientific validity and participant safety.
Institutional Review Board (IRB) / Independent Ethics Committee (IEC)
Formation and Ethical Oversight
The Institutional Review Board (IRB) or Independent Ethics Committee (IEC) plays a central role in protecting the rights, safety, and well-being of clinical trial participants. These committees are composed of medical experts, scientists, legal professionals, and lay members to ensure balanced decision-making.
Their responsibilities include reviewing trial protocols, informed consent documents, and investigator qualifications. They also monitor ongoing studies, review adverse events, and have the authority to suspend or terminate trials if ethical standards are compromised. In India, ethics committees operate under the oversight of the Central Drugs Standard Control Organization (CDSCO).
Informed Consent Process and Procedures
Ensuring Voluntary Participation
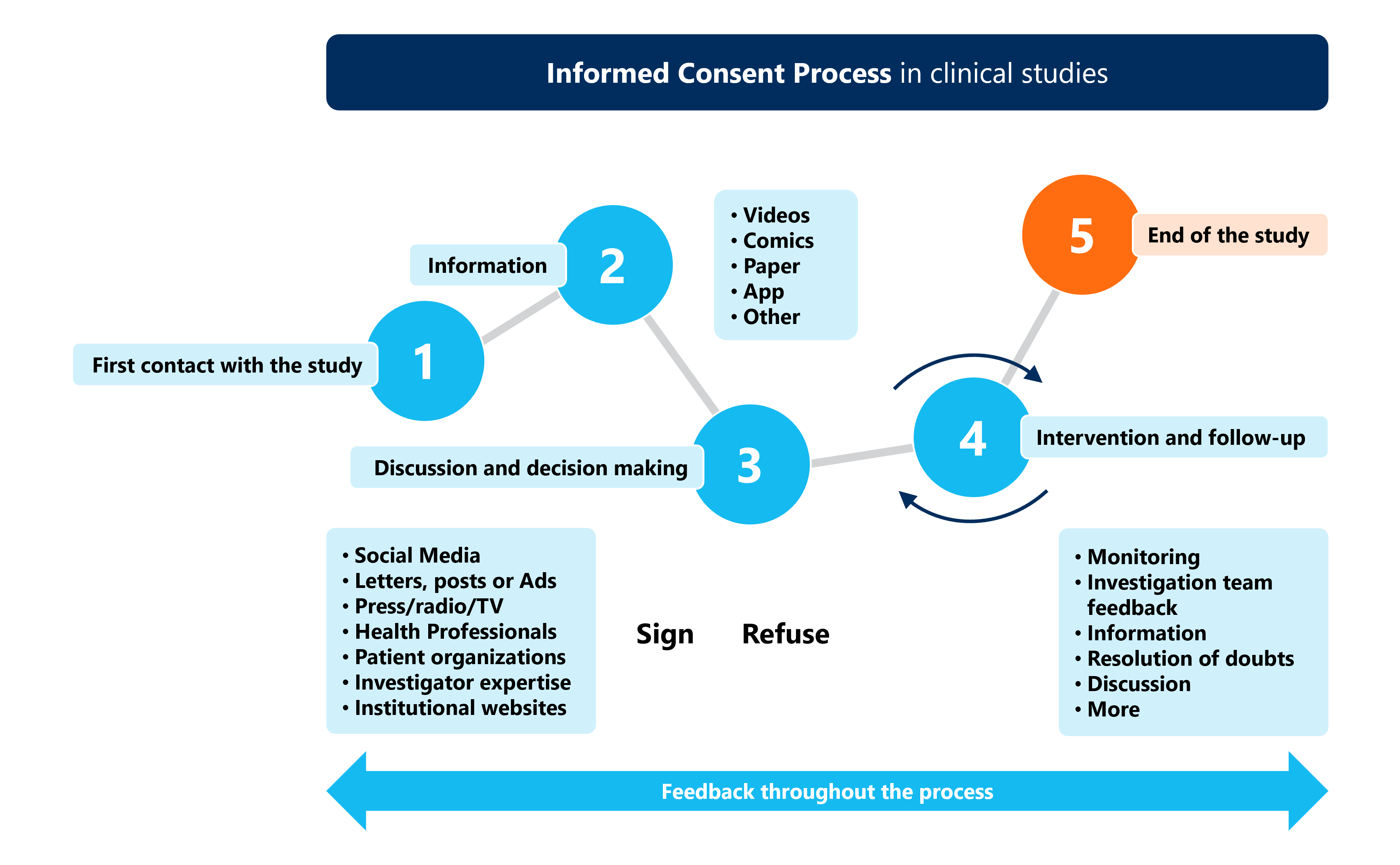
The informed consent process is a cornerstone of ethical clinical research. It ensures that participants voluntarily agree to take part in a trial after fully understanding its purpose, procedures, risks, benefits, and alternatives. Consent is not a one-time event but an ongoing process throughout the trial.
Investigators must provide information in a language understandable to participants and allow sufficient time for questions. Special safeguards are required for vulnerable populations, ensuring that consent is free from coercion or undue influence.
GCP Obligations of Investigators, Sponsors, and Monitors
Shared Responsibilities in Clinical Research
Good Clinical Practice (GCP) outlines the ethical and scientific quality standards for conducting clinical trials. Investigators are responsible for trial conduct at the site, ensuring participant safety, accurate data recording, and protocol compliance. They must maintain essential documents and promptly report adverse events.
Sponsors bear overall responsibility for trial initiation, management, financing, and quality assurance. They design protocols, select qualified investigators, and ensure proper monitoring. Clinical trial monitors act on behalf of sponsors to verify data accuracy, protocol adherence, and regulatory compliance through regular site visits and audits.
Globally, GCP principles are harmonized through guidelines issued by the International Council for Harmonisation and enforced by regulators such as the Food and Drug Administration (FDA) and the European Medicines Agency (EMA).
Managing and Monitoring Clinical Trials
Ensuring Quality and Compliance
Effective management and monitoring are essential to maintain trial integrity. Clinical trial management includes site coordination, data management, investigational product handling, and regulatory reporting. Monitoring activities focus on verifying informed consent, source data accuracy, and adherence to the approved protocol.
Risk-based monitoring approaches are increasingly adopted, allowing sponsors to focus resources on critical trial aspects. This improves efficiency while maintaining high-quality standards.
Pharmacovigilance: Safety Monitoring in Clinical Trials
Detecting and Managing Risks
Pharmacovigilance during clinical trials involves systematic monitoring, assessment, and reporting of adverse events and serious adverse events. Its primary goal is to identify potential safety concerns early and protect participants from harm.
Investigators must promptly report adverse events to sponsors and ethics committees. Sponsors, in turn, analyze safety data and submit periodic safety update reports to regulatory authorities. Robust pharmacovigilance systems contribute to informed risk–benefit assessments and guide decisions on trial continuation or modification.