
Pharmacovigilance has become a cornerstone of modern healthcare systems, ensuring that medicines remain safe throughout their lifecycle. While clinical trials provide essential safety data before approval, real-world use often reveals rare or long-term adverse effects. Pharmacovigilance bridges this gap by continuously monitoring, assessing, and preventing adverse drug reactions (ADRs), thereby safeguarding public health on a global scale.
Download UNIT 1 – Introduction to Pharmacovigilance — Protecting Patients Through Medicine Safety Notes
Get simplified revision notes for this unit:
⬇️
Download Unit 1 Notes PDF
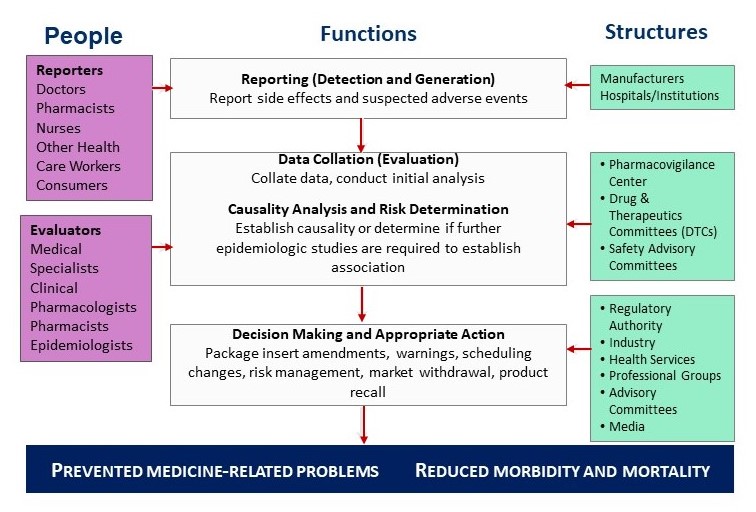
Concept and Scope of Pharmacovigilance
Pharmacovigilance is defined as the science and activities related to the detection, assessment, understanding, and prevention of adverse effects or any other medicine-related problems. Its scope extends beyond adverse drug reactions to include medication errors, lack of efficacy, drug interactions, misuse, and quality defects. By systematically collecting and analyzing safety data, pharmacovigilance supports informed regulatory decisions and safer clinical practice.
History and Development of Pharmacovigilance
From Drug Disasters to Structured Safety Systems
The formal development of pharmacovigilance was driven by tragic drug-related events, most notably the thalidomide disaster of the early 1960s, which caused severe birth defects. This incident highlighted the limitations of pre-marketing testing and prompted governments worldwide to establish post-marketing surveillance systems.
Over the decades, pharmacovigilance evolved into a structured scientific discipline, supported by national regulatory authorities and international collaboration. Advances in data management, epidemiology, and risk assessment have further strengthened its role in ensuring medicine safety.
Importance of Safety Monitoring of Medicines
Protecting Public Health in Real-World Use
Safety monitoring of medicines is essential because no drug is completely free from risk. Once a medicine is marketed, it is used by diverse populations, including children, elderly patients, and those with multiple comorbidities. Pharmacovigilance helps identify rare, delayed, or population-specific adverse effects that may not be detected during clinical trials.
Effective safety monitoring enables early detection of safety signals, timely risk communication, and regulatory actions such as label changes, restricted use, or product withdrawal. Ultimately, it ensures that the benefit–risk balance of medicines remains favorable.
WHO International Drug Monitoring Programme

Global Collaboration for Medicine Safety
The World Health Organization (WHO) established the International Drug Monitoring Programme to promote global cooperation in pharmacovigilance. The programme is coordinated through the Uppsala Monitoring Centre (UMC) in Sweden, which manages the global database of individual case safety reports.
Member countries contribute adverse drug reaction reports to this shared database, enabling detection of global safety signals and emerging risks. The WHO programme strengthens pharmacovigilance capacity worldwide, particularly in developing countries, by providing technical guidance, training, and standardized tools.
Pharmacovigilance Programme of India (PvPI)
Strengthening Medicine Safety in India
India’s national pharmacovigilance system is the Pharmacovigilance Programme of India (PvPI), launched to monitor adverse drug reactions across the country. PvPI is coordinated by the Indian Pharmacopoeia Commission and functions under the oversight of the Central Drugs Standard Control Organization (CDSCO).
PvPI operates through a network of Adverse Drug Reaction Monitoring Centres (AMCs) located in medical colleges and hospitals. These centres collect, analyze, and report ADR data, contributing to both national and global safety databases.
Introduction to Adverse Drug Reactions (ADRs)
Definitions and Classification of ADRs
An adverse drug reaction is defined as a harmful and unintended response to a medicine occurring at normal doses used for prophylaxis, diagnosis, or therapy. ADRs are commonly classified based on their mechanism, predictability, and severity. Classical classifications include dose-related reactions, idiosyncratic reactions, chronic effects, delayed reactions, and withdrawal reactions.
Understanding ADR classification helps healthcare professionals anticipate risks and implement appropriate preventive measures.
Detection and Reporting of ADRs
Role of Healthcare Professionals and Patients
Detection of ADRs relies on vigilant observation by healthcare professionals and, increasingly, patient self-reporting. Spontaneous reporting systems form the backbone of pharmacovigilance, allowing suspected ADRs to be reported even when a causal relationship is uncertain.
Timely and accurate reporting provides valuable data for signal detection and regulatory evaluation. Under-reporting remains a major challenge, making awareness and training essential components of effective pharmacovigilance.
Methods in Causality, Severity, and Risk Assessment
Assessing the Nature and Impact of ADRs
Causality assessment methods are used to evaluate the likelihood that a drug caused a reported adverse event. These methods consider factors such as temporal relationship, dechallenge and rechallenge, alternative causes, and existing evidence.
Severity assessment describes the intensity of an ADR, while seriousness refers to its clinical outcome, such as hospitalization, disability, or death. Predictability and preventability assessments help determine whether an ADR could have been anticipated or avoided, guiding risk minimization strategies.
Management of Adverse Drug Reactions
Clinical and Regulatory Responses
Management of ADRs involves prompt identification, discontinuation or modification of therapy, symptomatic treatment, and patient counseling. At the regulatory level, aggregated safety data may lead to updates in prescribing information, safety alerts, or regulatory restrictions.
Basic Terminologies Used in Pharmacovigilance
Standardizing Safety Communication
Pharmacovigilance relies on standardized terminology to ensure consistency in safety reporting and regulatory communication. Terms such as adverse event, serious adverse event, signal, risk management, and post-marketing surveillance are widely used across regulatory systems. Regulatory terminologies help align national and international pharmacovigilance practices, enabling effective global collaboration.